
HeartMate Recall: What You Need to Know
The recent announcement from the U.S. Food and Drug Administration’s recall of two heart devices has sparked concern throughout the medical community. Manufactured by Thoratec Corporation, a subsidiary of Abbott Laboratories, HeartMate II and HeartMate 3 have been associated with numerous injuries and at least 14 fatalities. The devices remain on the market despite these troubling findings, raising questions about the transparency and timing of reporting issues related to approved medical devices. This recall has left many individuals and families seeking clarity and assistance. At Ethen Ostroff Law, we recognize the complexities surrounding product recalls and stand ready to offer support and guidance to those impacted by the Heartmate recall.
HeartMate II and HeartMate 3
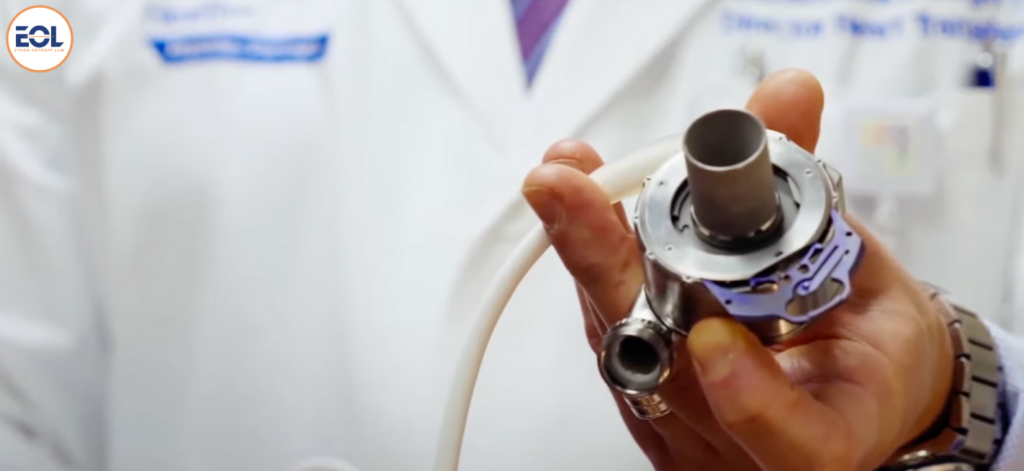
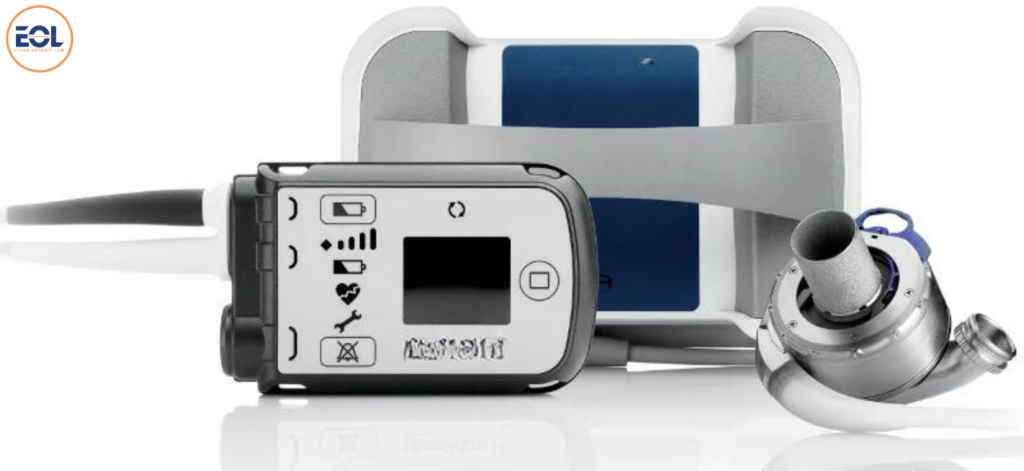
HeartMate II and HeartMate 3 are specialized left ventricular assist devices (LVADs) employed to aid patients with advanced heart failure, whether awaiting transplantation or ineligible for the procedure. HeartMate II, a longstanding continuous-flow LVAD, serves as a bridge to transplantation or as a long-term therapy option. In contrast, HeartMate 3, a more recent iteration, also utilizes continuous flow technology but integrates a fully magnetically levitated rotor to decrease the risk of device-related complications such as thrombosis and stroke.
These LVADs support the heart in pumping blood when its natural function is impaired, catering to adult patients with severe left ventricular heart failure for both short-term and long-term periods. They can be utilized while awaiting transplantation, facilitating cardiac recovery, or serving as a permanent solution in cases where transplantation is not feasible. HeartMate 3 extends its utility to pediatric patients as well. Functionally, both devices take over the pumping function of the left ventricle, redirecting blood from the weakened ventricle into the body’s main artery, the aorta, to ensure proper circulation. Their applications span both hospital and outpatient settings.

Abbott Recall 2024: HeartMate
In April 2024, the FDA issued a Class I recall for Abbott/Thoratec’s HeartMate II and HeartMate 3 devices, indicating a serious risk of injury or death. The recall was prompted by the accumulation of biological material in the devices, hindering their ability to assist the heart in circulating blood effectively.
The specific issue, known as Extrinsic Outflow Graft Obstruction (EOGO), occurs when biological material builds up between the HeartMate Outflow Graft and other components, impeding the device’s function. This buildup can trigger low blood flow alarms and diminish the device’s effectiveness in supporting the heart.
The recall affects a substantial number of devices, with around 14,000 units recalled in the US alone, and over 22,000 HeartMate 3 devices and more than 2,200 HeartMate II devices distributed globally. The devices remain on the market despite the recall. Reports have linked these devices to 273 injuries and at least 14 deaths, prompting urgent corrective action.
On February 19, 2024, Thoratec issued an Urgent Medical Device Correction Letter to affected customers, advising them to acknowledge receipt and pay attention to low flow alarms, which may indicate outflow graft obstruction. Abbott/Thoratec will continue to send letters to new recipients until corrective measures are implemented. Additionally, guidance on diagnosing and managing unresolved low flow associated with outflow graft obstruction was provided to customers.
List of HeartMate Devices
Here is the full list of affected devices, including their Unique Device Identifier (UDI) and Firm Reference Number for devices marketed in the US:
- HeartMate II Sealed Outflow Graft with Bend Relief
UDI-ID: 00813024010807
Firm Reference Number: 103393
- HeartMate II LVAS Implant Kit (Unsealed Outflow Graft with Bend Relief)
UDI-ID: 00813024011224
Firm Reference Number: 106015
- HeartMate II LVAS Implant Kit
UDI-ID: 00813024010005
Firm Reference Number: 1355
- HeartMate II LVAS Implant Kit (with RSOC Controller)
UDI-ID: 00813024010616
Firm Reference Number: 103695
- HeartMate II LVAS Implant Kit (with Sealed Graft)
UDI-ID: 00813024011170
Firm Reference Number: 104911
- HeartMate II LVAS Implant Kit (Used for pump exchange, No Graft)
UDI-ID: 00813024011996
Firm Reference Number: 107801
- HeartMate 3 Sealed Outflow Graft with Bend Relief
UDI-ID: 00813024013266
Firm Reference Number: 105581US
- HeartMate 3 LVAS Implant Kit
UDI-ID: 00813024013297
Firm Reference Number: 106524US
Complications Associated with HeartMate II and HeartMate 3
The recall of HeartMate II and HeartMate 3 devices poses serious risks to patients who have received these LVAS. The FDA warns of potential life-threatening blockages caused by the buildup of biological material in the devices. This risk can result in severe health complications, including:
- Death.
- Lightheadedness.
- Loss of consciousness.
- Refractory postural dizziness.
- Severe dizziness.
- Sudden changes in blood flow.
- Syncope (fainting).
Patients with these devices must promptly seek medical attention if they experience any symptoms or device-related issues. Healthcare providers play a crucial role in closely monitoring patients with HeartMate II and HeartMate 3 devices, taking appropriate actions to address potential obstructions or complications as they arise.
Who May be Affected
Those potentially affected include:
- Healthcare professionals who utilize the HeartMate II and HeartMate 3 LVAS to administer care.
- Individuals undergoing treatment with either the HeartMate II or HeartMate 3 Left Ventricular Assist Systems or LVAS.
Delays in Action: A Cause for Concern
The delayed recall of the HeartMate devices has sparked concerns about the reporting and handling of safety issues linked to medical devices. While certain surgeons were cognizant of potential problems with the devices, others, particularly those with less frequent implantation experience, may have been unaware. This knowledge gap could leave patients susceptible to adverse events.
Addressing the Issue
To address the issue, physicians are advised to monitor patients for “low-flow alarms” on the devices, signaling a potential obstruction. If such an obstruction is detected, physicians may choose to closely monitor the patient or conduct surgery to rectify the blockage. Abbott has provided assurance that rates of outflow obstruction are low, and patients with properly functioning devices need not be unduly concerned.
Past Recalls and Responses
The recent recall is not the first instance of issues being identified with HeartMate devices. Previous recalls have raised concerns, such as the potential twisting of the graft line carrying blood from the pump to the aorta, as noted in the May 2018 recall. This problem was specific to the HeartMate II device. It necessitated Abbott’s corrective action notices to hospitals and physicians to recall HeartMate II devices for further evaluation.
Moreover, in January 2024, Abbott issued an urgent correction letter to hospitals regarding a separate issue where the HeartMate 3 could inadvertently start and stop due to the pump’s communication system, utilized by cardiologists to assess patients’ status. The FDA publicly alerted about this matter in March.
In February 2024, Abbott issued another urgent letter to hospitals regarding the blockage issue, urging them to inform physicians, complete and return an acknowledgment form, and remain vigilant for low-flow alarms on the device’s monitor that may signal an obstruction. The company indicated in the letter that it is actively working on “a design solution” to mitigate blockages.
Future Implications
The latest recall carries significant implications, especially for patients with end-stage heart failure who depend on devices like HeartMate 3 and HeartMate II as their sole medical recourse. If the recall results in the withdrawal of these devices from the market, patients could find themselves without viable alternatives. This underscores the critical need for ongoing research and development in this area to ensure continued access to life-saving treatments.
What to Do If Impacted by the Abbott HeartMate Recall
If you or a loved one is affected by the Abbott HeartMate recall, follow these steps:
- Seek medical attention: See your doctor right away if you notice any symptoms or problems with your HeartMate device. Discuss your concerns with them.
- Document your treatment: Keep detailed records of your medical care related to your HeartMate device. This information can help your doctor and may be important for any legal matters.
- Consult a lawyer: Speak to a lawyer who understands product recalls and liability. They can explain your options and help you seek compensation if needed.
- Report issues: If you experience any problems with your HeartMate device, report them to MedWatch, the FDA’s program for medical device issues. You can do this online, by mail, or by fax.
- Stay updated: Stay informed about the Heartmate II and HeartMate 3 recall by following updates from the manufacturer and regulatory agencies. This will help you make informed decisions.
- Join support groups: Consider joining support groups for individuals affected by the HeartMate recall. Connecting with others can provide emotional support and valuable information.
- Take care of yourself: Dealing with a recall can be stressful, so prioritize your physical and emotional well-being. Seek support from loved ones or professionals if needed. Your health is important.
Process for Reporting a HeartMate Recall
Healthcare professionals and consumers are encouraged to report any adverse events or problems with medical devices, including HeartMate devices, to help the agency monitor their safety and effectiveness. To report any adverse reactions or quality issues related to HeartMate devices to MedWatch: The FDA Safety Information and Adverse Event Reporting Program, follow these steps:
Information to Provide When Reporting a HeartMate Device Recall
When reporting a HeartMate device recall, ensure you provide the following information:
- Device information: Specify the type of HeartMate device you have (HeartMate II or HeartMate 3). Include relevant model numbers or serial numbers.
- Issue details: Describe the specific problem or reason for the recall you are experiencing with the device. Mention any symptoms or complications observed.
- Patient information: Provide your personal details, such as name, contact information, and medical history. If reporting for someone else, include their information.
- Healthcare provider details: Include the name and contact details of the healthcare provider who implanted or manages the device.
- Date and circumstances: Mention when the issue was noticed and any relevant circumstances surrounding it.
- Action taken: Describe any steps you have already taken regarding the device issue.
By providing accurate and comprehensive information, you contribute to addressing the recall effectively and ensuring patient safety.
Process for Patients to Return Recalled HeartMate Devices
Here is the process for patients to return recalled Abbott HeartMate 3 and HeartMate II devices:
- Contact Abbott: Patients should reach out to Abbott Technical Support at 1-800-456-1477 for guidance on returning the recalled devices. US customers with questions about the recall should contact Abbott/Thoratec at (844) 692-6367.
- Follow Abbott’s instructions: Patients must adhere to any specific instructions provided by Abbott regarding the return process for the recalled HeartMate II and HeartMate 3 devices.
- Provide necessary information: Patients may need to furnish device-specific details, such as model numbers or serial numbers, to facilitate the return process efficiently.
- Return the devices: Patients should carefully package the recalled HeartMate II and HeartMate 3 devices as per Abbott’s instructions and arrange for their return to the designated location.
By following these steps and communicating with Abbott Technical Support, patients can ensure the safe and proper return of the recalled HeartMate II and HeartMate 3 devices.
Seeking Legal Guidance
If you or someone you care about has been impacted by the HeartMate recall, seeking legal guidance is essential. Understanding your rights and exploring compensation options can be crucial during this challenging time.
Why You Need Ethen Ostroff Law
In the wake of the Heartmate recall, uncertainty can be overwhelming for individuals and families. However, with informed decision-making and legal support, navigating this challenging time is possible. Ethen Ostroff Law specializes in aiding those impacted by product recalls and defective medical devices. Our expertise in product liability and medical device recalls enables us to offer guidance and assistance throughout the HeartMate recall process. Our seasoned attorneys will evaluate your case, outline your legal options, and advocate for your rights to seek compensation for any injuries or losses resulting from the faulty HeartMate device. With Ethen Ostroff Law, your rights are safeguarded.
What are you waiting for? Contact Ethen Ostroff Law now at 610-510-8883 ( by calling this number, you consent to receive SMS updates from Ethen Ostroff Law) or Submit Form to get free consultation.
Related Questions on Abbott Heartmate Recall
What are the complications of HeartMate?
Complications of HeartMate devices may include infection, device malfunction, or blockages, which can hinder the device’s ability to circulate blood effectively.
Is HeartMate II being discontinued?
There have been no official announcements regarding the discontinuation of HeartMate II. However, the Abbott Heartmate recall has raised concerns about the future availability and safety of these devices.
Is HeartMate 3 FDA-approved?
Yes, Abbott HeartMate 3 received FDA approval in 2017. It is currently one of the primary medical options for patients with end-stage heart failure who are not eligible for a transplant.
Why was HeartWare discontinued?
The HeartWare HVAD, a ventricular assist device aiding heart failure patients, was discontinued due to safety concerns unrelated to the recent recall. Between May 2013 and June 2021, it was subject to 25 FDA recalls, with 60% classified as Class I, signifying serious injury or death risk. Over 100 complaints reported HVAD pump delay or failure to restart. Abbott, the manufacturer, might have halted production due to product issues or strategic business decisions.

